Иларис в Тамбове


Аналоги Иларис








Инструкция на Иларис
Выбор формы выпуска
Состав
1 мл препарата содержит:
действующее вещество: канакинумаб 150,00 мг;
вспомогательные вещества: маннитол, гистидин/гистидина гидрохлорида моногидрат, полисорбат-80, вода для инъекций.
Описание
От бесцветного до светло-коричневато-желтого цвета прозрачный или опалесцирующий раствор.
Фармакотерапевтическая группа
Иммунодепрессивное средство – антитела моноклональные к интерлейкину-1β.
Фармакологическое действие
Механизм действия
Канакинумаб – человеческое моноклональное антитело IgGl/каппа изотипа к интерлейкину-1β (ИЛ-1β). Канакинумаб с высоким аффинитетом связывается с человеческим ИЛ-1β, нейтрализуя, таким образом, его биологическое действие, блокируя взаимодействие ИЛ-1β с его рецепторами, ИЛ-1β-индуцированную активацию генов и продукцию медиаторов воспаления, таких как ИЛ-6 и циклооксигеназа-2. Таким образом, канакинумаб подходит для коррекции заболеваний и состояний, характеризующихся гиперпродукцией ИЛ-1β на местном или системном уровне.
Фармакодинамика
Избыточная продукция ИЛ-1β при воспалительных заболеваниях может приводить к развитию местного или системного воспалительного процесса, избыточной продукции С-реактивного белка (СРБ) или сывороточного амилоида А (САА) и лихорадки.
Криопирин-ассоциированный периодический синдром (cryopyrin-associated periodic syndrome, CAPS)
У пациентов с различными фенотипами CAPS, включая семейный холодовой аутовоспалительный синдром/семейную холодовую крапивницу (Familial Cold Autoinflammatory Syndrome/ Familial Cold Urticaria, FCAS/FCU), Синдром Макла-Уэлъса (Muckle-Wells Syndrome, MWS) и мультисистемное младенческое воспалительное заболевание/хронический младенческий неврологический кожно-артикулярный синдром (Neonatal onset multisystemic inflammatory disease/Chronic infantile neurological cutaneous and articular syndrome, NOMID/C1NCA), с избыточной неконтролируемой продукцией ИЛ-1β (что проявляется лихорадкой, повышенной утомляемостью, кожной сыпью, артритом, выраженным лейкоцитозом, большим количеством тромбоцитов, а также увеличением концентрации СРБ) отмечен быстрый ответ на терапию канакинумабом. На фоне терапии препаратом такие явления как повышенная концентрация СРБ, САА, лейкоцитоз и повышенное количество тромбоцитов быстро возвращались к нормальному уровню.
Болезнь Стилла у взрослых и юношеский артрит с системным началом (системный ювенильный идиопатический артрит, сЮИА, классификация ILAR, международной лиги ревматологических ассоциаций)
Болезнь Стилла у взрослых и сЮИА – тяжелые аутовоспалительные заболевания, которые обусловлены врожденным иммунитетом посредством провоспалительных цитокинов, ключевым среди которых является ИЛ-1β.
Общие проявления болезни Стилла взрослых и сЮИА включают лихорадку, кожную сыпь, гепатоспленомегалию, лимфаденопатию, полисерозит и артрит. Лечение препаратом у большинства данных пациентов приводило к быстрому и устойчивому улучшению со стороны суставных и системных проявлений сЮИА со значительным уменьшением количества воспаленных суставов, быстрому разрешению лихорадки и уменьшению концентрации реактантов острой фазы.
Подагрический артрит
Обострение подагрического артрита обусловлено активацией тканевых макрофагов и одновременной гиперпродукцией ИЛ-1β, что приводит к острому воспалительному ответу с выраженным болевым синдромом. Продукция ИЛ-1β провоцируется отложением кристаллов солей мочевой кислоты (мононатрия урат моногидрата) в суставах и окружающих тканях, что приводит к активации комплексов «NALP3-инфламосома». Такие активаторы врожденного иммунитета, как эндогенные агонисты толл-подобных рецепторов, также могут вносить свой вклад в активацию транскрипции гена ИЛ-1β, инициируя обострение подагрического артрита. После терапии канакинумабом в течение короткого времени отмечается снижение концентрации лабораторных маркеров воспаления (СРБ, САА), и исчезают признаки воспаления пораженного сустава (боль, отек, покраснение).
Фармакокинетика
Всасывание
У взрослых пациентов с различными фенотипами CAPS после однократного подкожного (п/к) введения 150 мг канакинумаба время достижения максимальной концентрации (Сmax) составляет около 7 дней. Средний конечный период полувыведения составляет 26 дней. При п/к введении канакинумаба абсолютная биодоступность составляет 66% (популяционный фармакокинетический анализ у пациентов с CAPS, включая детей в возрасте от 2 лет). Параметры фармакокинетики (площадь под кривой «концентрация – время» (AUC) и Сmax) повышаются пропорционально дозе в диапазоне доз от 0,30 до 10,0 мг/кг при внутривенной (в/в) инфузии или при п/к введении (в дозе от 150 до 600 мг).
Распределение
Канакинумаб связывается с сывороточным ИЛ-1β. Объем распределения (Vss) изменяется в зависимости от массы тела.
У пациентов с CAPS Vss составляет 6,2 л при массе тела 70 кг, 5,0 л – у пациентов с синдромом периодической лихорадки (TRAPS, H1DS/MKD, FMF) с массой тела 55 кг, у пациентов с сЮИА – 3,2 л при массе тела 33 кг и у пациентов с подагрическим артритом – 7,9 л при массе тела 93 кг. При п/к введении препарата в течение 6 месяцев в дозе 150 мг каждые 8 недель, в дозе 4 мг/кг каждые 4 недели, 150 мг каждые 12 недель коэффициент кумуляции канакинумаба составляет 1,3, 1,6 и 1,1 соответственно.
Выведение
Клиренс (С1) изменяется в зависимости от массы тела. Для пациентов с CAPS этот показатель составляет 0,17 л/сут при массе тела 70 кг, 0,14 л/сут – у пациентов с синдромом периодической лихорадки (TRAPS, HIDS/MKD, FMF) при массе тела 55 кг, у пациентов с сЮИА – 0,11 л/сут при массе тела 33 кг, у пациентов с подагрическим артритом – 0,23 л/сут при массе тела 93 кг. После учета весовых различий существенной разницы в фармакокинетических свойствах канакинумаба у пациентов с подагрическим артритом, различными фенотипами CAPS, TRAPS, HIDS/MKD, FMF и сЮИА выявлено не было. При повторном применении препарата не наблюдается увеличения С1 или изменений каких-либо других, зависимых от времени фармакокинетических параметров канакинумаба. После коррекции дозы по массе тела не отмечено изменения фармакокинетических показателей в зависимости от возраста и пола.
Фармакокинетика в особых клинических случаях
Пациенты в возрасте младше 18 лет
У пациентов в возрасте от 4 лет и старше после однократного п/к введения препарата в дозе 150 мг или 2 мг/кг 1 раз время достижения Сmax канакинумаба составляет 2-7 дней.
Конечный период полувыведения канакинумаба у пациентов данной категории сходен с таковым у взрослых и составляет от 22,9 до 25,7 дней.
По данным анализа популяционного фармакокинетического моделирования фармакокинетические параметы канакинумаба у детей с CAPS в возрасте от 2 до 4 лет сходны с таковыми у пациентов 4 лет и старше. Дополнительный фармакокинетический анализ показал, что параметры фармакокинетики канакинумаба у 6 детей с CAPS в возрасте младше 2 лет были сходными с таковыми у детей в возрасте старше 2 лет. Фармакокинетические характеристики у пациентов с CAPS, TRAPS, HIDS/MKD, FMF и сЮИА схожи. При п/к введении канакинумаба в дозе 4 мг/кг каждые 4 педели у пациентов с сЮИА значения AUC и Сmax были аналогичны в возрастной группе от 2 до 20 лет.
По данным анализа популяционного фармакокинетического моделирования фармакокинетические параметры канакинумаба у пациентов с сЮИА в возрасте 16-20 лет и возрасте младше 16 лет были схожими. У пациентов с сЮИА в возрасте старше и младше 20 лет расчетная экспозиция канакинумаба в равновесном состоянии при величине дозы 4 мг/кг (максимум 300 мг) были сопоставимы.
У пациентов с синдромом периодической лихорадки (TRAPS, HIDS/MKD, FMF) экспозиция при минимальной концентрации канакинумаба в плазме крови при п/к введении препарата в дозе 2 мг/кг каждые 4 недели сопоставима в возрастных группах у пациентов в возрасте от 2 до 20 лет.
Пациенты в возрасте старше 65 лет
Не было выявлено разницы в фармакокинетических параметрах, основанных на С1 и Vss, у пациентов старшей возрастной группы и пациентов в возрасте менее 65 лет.
Иларис: Показания
Аутовоспалителъные синдромы периодической лихорадки:
- криопирин-ассоциированный периодический синдром (CAPS) у взрослых и детей в возрасте 2 лет и старше, включая:
- семейный холодовой аутовоспалительный синдром (РСА8)/семейная холодовая крапивница (FCU),
- синдром Макла-Уэльса (MWS),
- младенческое мультисистемное воспалительное заболевание (NOMID)/хронический младенческий неврологический кожно-артикулярный синдром (CINCA);
- периодический синдром, ассоциированный с рецепторами к фактору некроза опухоли (TRAPS) у взрослых и детей в возрасте 2 лет и старше;
- гипер-IgD-синдром/синдром дефицита мевалонат-киназы (HIDS/MKD) у взрослых и детей в возрасте 2 лет и старше;
- семейная средиземноморская лихорадка (FMF) у взрослых и детей в возрасте 2 лет и старше в монотерапии при наличии противопоказаний к /непереносимости терапии колхицином или в комбинации с колхицином при отсутствии адекватного терапевтического ответа на монотерапию максимально переносимой дозой колхицина.
Активная фаза болезни Стилла, в том числе болезни Стилла взрослых (БСВ) и системного ювенильного идиопатического артрита (сЮИА) у пациентов от 2 лет и старше при неадекватном ответе на терапию нестероидными противовоспалительными препаратами (НПВП) и системными глюкокортикостероидными препаратами. Препарат Иларис® можно применять в монотерапии и в комбинации с метотрексатом.
Острый подагрический артрит с целью лечения частых острых приступов подагрического артрита и предупреждения развития новых приступов при неэффективности, непереносимости или при наличии противопоказаний к применению нестероидных противовоспалительных препаратов и/или колхицина и при невозможности проведения терапии повторными курсами глюкокортикостероидов.
Способ применения и дозы
У пациентов с аутовоспалительными синдромами периодических лихорадок (CAPS, TRAPS, HIDS/MKD, FMF), а также болезнью Стилла, в том числе с сЮИА терапия препаратом Иларис® может быть инициирована и проводиться только под контролем врача, имеющего опыт диагностики и лечения соответствующих заболеваний. После обучения технике приготовления раствора, выбору шприцов и игл, пригодных для инъекции, а также технике проведения п/к инъекции, пациенты с вышеуказанными заболеваниями или лица, за ними ухаживающие, могут самостоятельно вводить препарат под надлежащим контролем (если врач сочтет это необходимым).
У пациентов с подагрическим артритом терапия препаратом Иларис® может быть инициирована и проводиться только под контролем врача, имеющего опыт применения биологических лекарственных препаратов; введение препарата должно осуществляться только медицинским работником.
Препарат вводят п/к, выбирая участки тела с выраженной подкожной клетчаткой, например, передняя поверхность живота, передняя поверхность верхней части бедра, наружная поверхность верхней части плеча или ягодицы (см. раздел «Указания по применению»). Во избежание болезненных ощущений следует менять место введения при каждой последующей инъекции. Следует избегать введения препарата в участки с нарушением целостности кожных покровов, области ушибов или с наличием сыпи. Следует избегать введения препарата в рубцовую ткань в связи с возможным снижением экспозиции канакинумаба.
Каждый флакон препарата Иларис® предназначен для однократного использования у одного пациента для введения однократной дозы.
Криопирин-ассоциированный периодический синдром (CAPS)
Рекомендованные начальные дозы препарата Иларис® для пациентов с криопирин-ассоциированным периодическим синдромом указаны в таб. 1
Таб.1 Рекомендованные начальные дозы препарата Иларис® для пациентов с криопирин-ассоциированным периодическим синдромом
| Категория пациентов | Масса тела | Рекомендованная начальная доза |
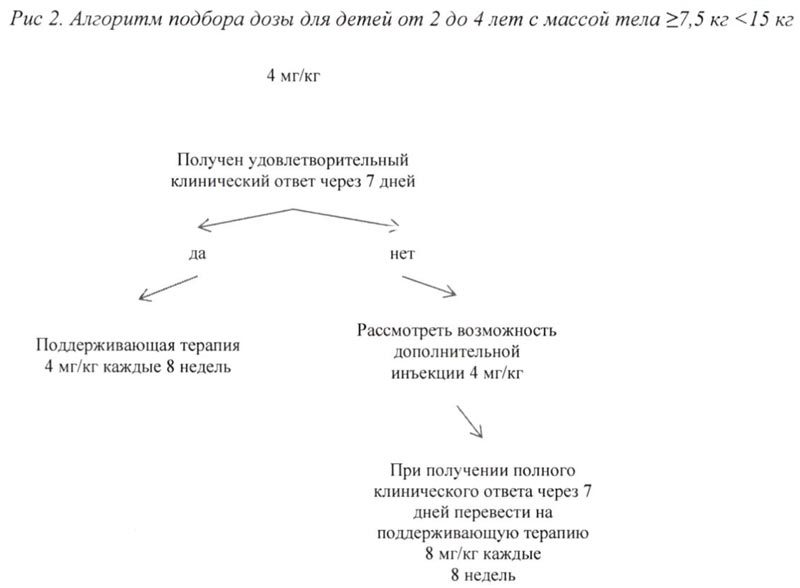
| Дети в возрасте от 2 лет до 4 лет | ≥7,5 кг | 4 мг/кг |
| Взрослые и дети старше 4 лет | >40 кг | 150 мг |
| ≥15 - ≤40 кг | 2 мг/кг | |
| ≥7,5 - <15 кг | 4 мг/кг |
Препарат вводят п/к, 1 инъекция с интервалом 8 недель.
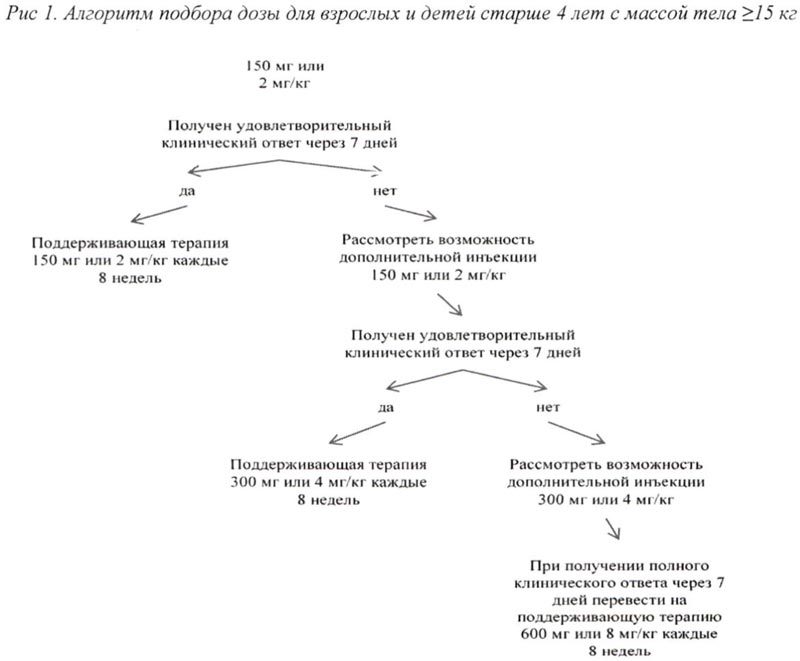
Если при стартовой дозе 150 мг или 2 мг/кг не получен удовлетворительный клинический ответ, а именно процесс разрешения сыпи и других симптомов воспаления не наблюдается в течение 7 дней после первой инъекции препарата Иларис®, возможно проведение второй инъекции препарата в дозе 150 мг (при массе тела >40 кг) или 2 мг/кг (при массе тела ≥15 кг и ≤40 кг). При достижении в последующем полного клинического ответа данным пациентам рекомендовано проводить поддерживающую терапию препаратом Иларис® в дозе 300 мг 1 инъекция с интервалом 8 недель (при массе тела >40 кг) или 4 мг/кг 1 инъекция с интервалом 8 недель (при массе тела ≥15 кг и ≤40 кг).
Если удовлетворительный клинический эффект не наблюдается в течение 7 дней после повышения дозы, возможно проведение третьей инъекции препарата Иларис® в дозе 300 мг (при массе тела >40 кг) или 4 мг/кг (при массе тела ≥15 кг и ≤40 кг).
При достижении в последующем полного клинического ответа данным пациентам рекомендовано проводить поддерживающую терапию препаратом Иларис®в дозе 600 мг 1 инъекция с интервалом 8 недель (при массе тела >40 кг) или 8 мг/кг (при массе тела ≥15 кг и ≤40 кг) 1 инъекция с интервалом 8 недель на основании индивидуальной клинической оценки.
Если при стартовой дозе 4 мг/кг удовлетворительный клинический эффект не наблюдается в течение 7 дней после первой инъекции, возможно проведение второй инъекции препарата Иларис® в дозе 4 мг/кг. При достижении в последующем полного клинического ответа данным пациентам следует рассмотреть возможность поддерживающей терапии в дозе 8 мг/кг 1 инъекция с интервалом 8 недель на основании индивидуальной клинической оценки.
Алгоритм подбора дозы для взрослых и детей старше 4 лет с массой тела ≥ 15 кг представлен на рис. 1, для детей от 2 до 4 лет с массой тела ≥7,5 кг <15 кг представлен на рис. 2.
Клинический опыт применения препарата с интервалом дозирования менее 4 недель или в дозе более 600 мг или 8 мг/кг ограничен.
Периодический синдром, ассоциированный с рецепторами к фактору некроза опухоли (TRAPS), гипер-IgD-синдром/синдром дефицита мевалонат-киназы (HIDS/MKD), семейная средиземноморская лихорадка (FMF)
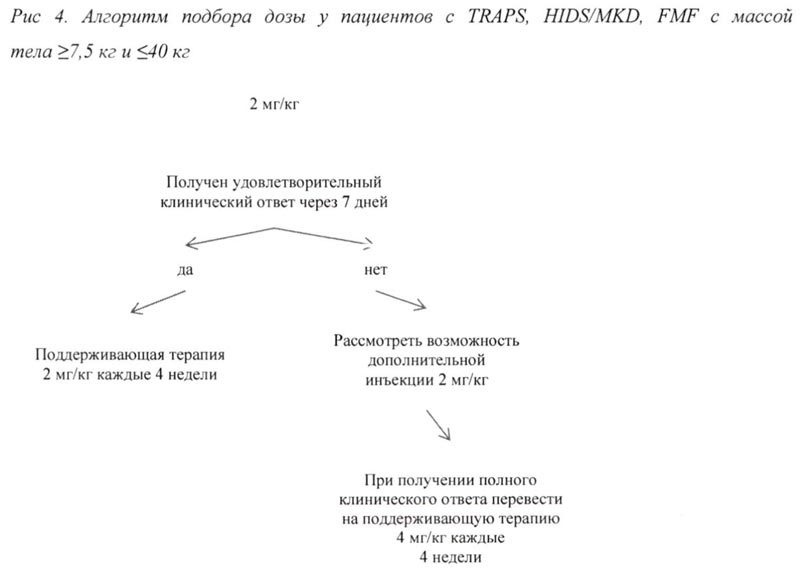
Рекомендованная начальная доза препарата у взрослых и детей в возрасте 2 лет и старше составляет:
- 150 мг у пациентов с массой тела >40 кг;
- 2 мг/кг у пациентов с массой тела ≥7,5 кг и ≤40 кг.
Препарат применяют в виде п/к инъекции каждые 4 недели.
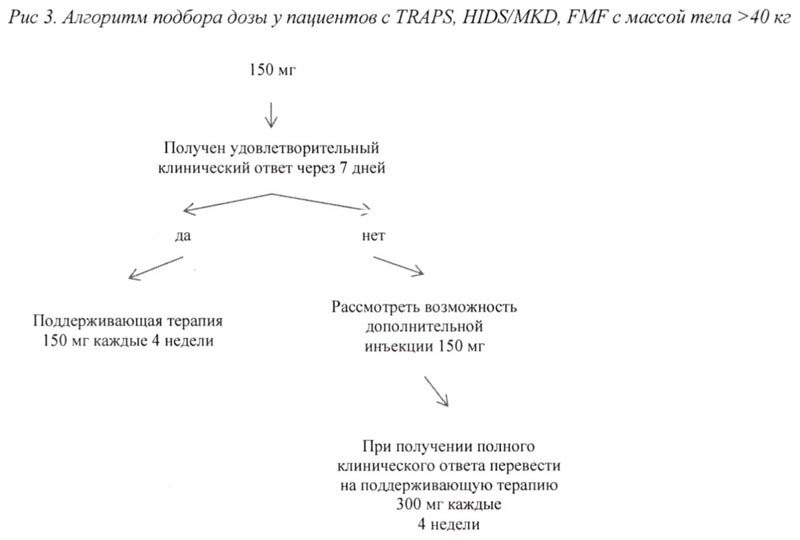
При отсутствии удовлетворительного клинического ответа в течение 7 дней после первой инъекции препарата Иларис® возможно проведение второй инъекции препарата в дозе 150 мг (при массе тела >40 кг) или 2 мг/кг (при массе тела ≥7,5 кг и ≤40 кг). При достижении в последующем полного клинического ответа данным пациентам рекомендовано проводить поддерживающую терапию препаратом Иларис® в дозе 300 мг или 4 мг/кг каждые 4 недели в виде п/к инъекции.
Продолжение терапии препаратом у пациентов без клинического улучшения должно быть повторно рассмотрено лечащим врачом.
Болезнь Стилла (болезнь Стилла у взрослых и сЮИА у пациентов в возрасте старше 2 лет)
Рекомендованная доза у пациентов с массой тела ≥7,5 кг составляет 4 мг/кг (максимально до 300 мг) каждые 4 недели в виде п/к инъекции. Продолжение терапии препаратом у пациентов без клинического улучшения должно быть повторно рассмотрено лечащим врачом.
Подагрический артрит
Контроль гиперурекимии и его оптимизацию следует проводить с помощью соответствующих противоподагрических препаратов.
Применять препарат Иларис® для терапии приступов обострения подагрического артрита следует по мере необходимости.
Рекомендованная доза препарата у взрослых составляет 150 мг, препарат вводят однократно п/к во время приступа подагрического артрита. Для достижения максимальной эффективности препарат необходимо вводить как можно раньше после начала приступа подагрического артрита.
Пациентам с отсутствием терапевтического ответа на первую инъекцию не следует вводить препарат повторно. У пациентов с положительным ответом на терапию препаратом при необходимости продолжения лечения повторное введение препарата возможно не ранее чем через 12 недель после предыдущей инъекции.
Применение у особых групп пациентов
Пациенты в возрасте старше 65 лет
Не требуется коррекции дозы препарата у пациентов в возрасте старше 65 лет.
Пациенты в возрасте младше 18 лет
Нет данных по применению препарата Иларис® для лечения подагрического артрита у детей и подростков младше 18 лет.
Опыт применения канакинумаба у детей младше 2 лет ограничен по показаниям криопирин-ассоциированный периодический синдром (CAPS), периодический синдром, ассоциированный с рецепторами к фактору некроза опухоли (TRAPS), гипер-IgD-синдром/синдромом дефицита мевалонат-киназы (HIDS/MKD), семейная средиземноморская лихорадка (FMF), доступные данные указаны в разделах «Фармакологические свойства» и «Побочное действие», однако рекомендации по режиму дозирования отсутствуют. Нет данных по эффективности и безопасности канакинумаба у пациентов с сЮИА младше 2 лет.
Пациенты с нарушением функции почек
Не требуется коррекции дозы препарата у пациентов с нарушениями функции почек (опыт клинического применения у таких пациентов ограничен).
Пациенты с нарушением функции печени
У пациентов с нарушениями функции печени эффективность и безопасность применения препарата не изучались. Так как канакинумаб является человеческим IgG, предполагается, что нарушение функции печени не влияет на его фармакокинетику.
Применение при беременности и кормлении грудью
Пациенты и пациентки с сохраненным репродуктивным потенциалом /контрацепция
Женщинам следует применять надежные методы контрацепции во время терапии препаратом Иларис® и в течение 3 месяцев после применения последней дозы препарата.
Беременность
Данные по применению канакинумаба у беременных пациенток ограничены. В исследованиях у животных не выявлено какой-либо репродуктивной токсичности. Риск для плода/матери неизвестен. В связи с вышесказанным применение препарата у беременных пациенток или у пациенток, планирующих беременность, возможно только после тщательной оценки отношения польза-риск.
В исследованиях у животных показано, что канакинумаб проникает через плаценту и обнаруживается у плода. Данные у человека отсутствуют, однако поскольку канакинумаб является иммуноглобулином класса G (IgGl) ожидается трансплацентарная передача у человека. Клиническое влияние данного явления неизвестно. Тем не менее, не рекомендовано применение живых вакцин у новорожденного, подверженного действию канакинумаба in utero, в течение 16 недель после применения последней дозы препарата у матери до родоразрешения.
Пациенток, получавших препарат во время беременности, следует проинформировать о необходимости сообщить о данном факте медицинскому работнику до проведения какой-либо вакцинации новорожденного.
Грудное вскармливание
Неизвестно, проникает ли канакинумаб в грудное молоко. Таким образом, решение о грудном вскармливании на фоне терапии препаратом следует принимать только после тщательной оценки отношения польза-риск.
В исследованиях у животных антимышиные ИЛ-1β-антитела передавались с молоком потомству и не оказывали какого-либо неблагоприятного воздействия на его развитие.
Фертильность
Официальные исследования для оценки возможного влияния препарата Иларис® на фертильность у человека не проводились. По данным исследований у самцов обезьян (Callithrix jacchus) канакинумаб не оказывает влияния на мужскую фертильность. Антимышиные ИЛ-1β-антитела не оказывают нежелательного влияния фертильность самок и самцов мышей.
Иларис: Противопоказания
- Подтвержденная повышенная чувствительность к действующему веществу или другим компонентам препарата.
- Острые тяжелые инфекционные заболевания.
- Возраст менее 2 лет (безопасность и эффективность для пациентов указанной категории изучены недостаточно).
С осторожностью
Препарат следует с осторожностью применять у пожилых пациентов; у пациентов с рецидивирующими инфекциями или любыми состояниями в анамнезе, предрасполагающими к развитию инфекций.
Иларис: Побочные действия
При применении препарата Иларис® в ходе клинических исследований отмечалось повышение частоты инфекционных заболеваний, преимущественно инфекций верхних дыхательных путей. Течение инфекционных заболеваний чаще всего было легкой или средней степени тяжести, однако отмечались и случаи тяжелого течения. На фоне терапии препаратом отмечены случаи развития реакций гиперчувствительности. Отмечены случаи развития оппортунистических инфекций на фоне лечения препаратом Иларис®.
Влияния продолжительной терапии на тип или частоту развития нежелательных лекарственных реакций (НЛР) не наблюдалось.
Нежелательные реакции (HP) сгруппированы в соответствии с классификацией органов и систем органов MedDRA, перечислены в порядке уменьшения важности. Частота встречаемости оценивалась следующим образом: очень часто (≥1/10); часто (≥1/100, <1/10); нечасто (≥1/1000, <1/100); редко (≥1/10000, <1/1000); очень редко (<1 /10000), частота неизвестна – недостаточно данных для оценки частоты развития.
CAPS, TRAPS, HIDS/MKD, FMF, сЮИА и подагрический артрит
Инфекционные и паразитарные заболевания: очень часто – инфекции дыхательных путей (в том числе пневмония, бронхит, грипп, вирусные инфекции, синусит, ринит, фарингит, тонзиллит, назофарингит, инфекции верхних дыхательных путей), инфекции уха, целлюлит, гастроэнтерит, инфекции мочевыводящих путей; часто – вульвовагинальный кандидоз.
Нарушения со стороны нервной системы, часто – головокружение/вертиго.
Нарушения со стороны желудочно-кишечного тракта: очень часто – боль в верхней части живота1; нечасто – гастроэзофагеальная рефлюксная болезнь2.
Нарушения со стороны кожи и подкожных тканей: очень часто – реакция в месте введения.
Нарушения со стороны скелетно-мышечной и соединительной ткани: очень часто – артралгия1; часто – скелетно-мышечная боль, боль в спине2.
Общие расстройства и нарушения в месте введения: часто – общая слабость/повышенная утомляемость2.
Лабораторные и инструментальные данные: очень часто – снижение почечного клиренса креатинина1,3, протеинурия1,4, лейкопения1,5; часто – нейтропения5; нечасто – уменьшение количества тромбоцитов5.
Ссылки:
1 – при СЮИА;
2 – при подагрическом артрите;
3 – исходя из расчетного клиренса креатинина, большинство случаев носили преходящий характер;
4 – в большинстве случаев при помощи тест-полоски определялись «следы белка» или «1+»;
5 – см. информацию ниже.
Описание отдельных нежелательных реакции
Долгосрочные данные и изменения лабораторных показателей у пациентов с CAPS
При применении препарата в клинических исследованиях у пациентов с CAPS отмечалось увеличение средних показателей концентрации гемоглобина и снижение количества лейкоцитов, нейтрофилов и тромбоцитов.
В редких случаях у пациентов с CAPS, получавших лечение препаратом, наблюдалось повышение активности «печеночных» трансаминаз.
В ряде случаев на фоне терапии препаратом у пациентов с CAPS отмечалось бессимптомное незначительное увеличение концентрации билирубина в сыворотке крови, не сопровождающееся повышением активности «печеночных» трансаминаз.
В долгосрочных открытых клинических исследованиях с эскалацией дозы случаи инфекций (гастроэнтерит, инфекция дыхательных путей, инфекция верхних дыхательных путей), рвота и головокружение более часто отмечались в группе 600 мг или 8 мг/кг по сравнению с группами, получавшими другую дозу.
Лабораторные показатели у пациентов с TRAPS, HIDS/MKD и FMF
Нейтрофилы
Несмотря на то, что уменьшение количества нейтрофилов ≥2 степени тяжести отмечено у 6,5% пациентов (часто) и уменьшение 1 степени тяжести отмечено у 9,5% пациентов, данные явления в целом носили преходящий характер; инфекция, ассоциированная со снижением количества нейтрофилов, не была определена как HP.
Тромбоциты
Несмотря на то, что уменьшение количества тромбоцитов (≥2 степени тяжести) отмечено у 0,6% пациентов, кровотечение не было определено как HP. Незначительное и транзиторное уменьшение количества тромбоцитов 1 степени тяжести отмечено у 15,9% пациентов без ассоциации с кровотечением.
Лабораторные показатели у пациентов с сЮИА
Гематологические показатели
Снижение количества лейкоцитов ≤0,8×нижней границы нормы (НГН) отмечалось у 33 пациентов (16,5%).
В программе клинической разработки при сЮИА транзиторное уменьшение абсолютного количества нейтрофилов до <1×109/л отмечено у 12 пациентов (6,0%).
В программе клинической разработки при сЮИА транзиторное уменьшение тромбоцитов <НГН отмечено у 19 пациентов (9,5%).
Аланинаминотрансфераза (АЛТ) и/или аспартатаминотрансфераза (ACT)
В программе клинической разработки при сЮИА увеличение АЛТ и/или АСТ>3×ВГН (высшей границы нормы) отмечено у 19 пациентов (9,5%).
Лабораторные показатели у пациентов с подагрическим артритом
Гематологические показатели
Снижение количества лейкоцитов ≤0,8×НГН отмечалось у 6,7% пациентов, получавших лечение препаратом Иларис® по сравнению с 1,4% пациентов, получавших триамцинолон. В сравнительных исследованиях снижение абсолютного количества нейтрофилов ниже 1×109/л отмечалось у 2% пациентов с подагрическим артритом. Наблюдались также изолированные случаи снижения количества нейтрофилов ниже 0,5×109/л.
В 12,7% случаев на фоне терапии канакинумабом наблюдалось легкое (<НГН и >75×109/л) и преходящее снижение количества тромбоцитов (у препарата сравнения данное снижение наблюдалось в 7,7% случаев).
Мочевая кислота
В сравнительных исследованиях у пациентов с подагрическим артритом после лечения препаратом Иларис® наблюдалось повышение концентрации мочевой кислоты (0,7 мг/дл на 12 неделе и 0,5 мг/дл на 24 неделе). В другом исследовании у пациентов, начавших лечение противоподагрическими препаратами, повышения концентрации мочевой кислоты не наблюдалось. Повышение концентраций мочевой кислоты в ходе клинических исследований у пациентов с неподагрическим артритом не наблюдалось.
АЛТ/АСТ
К концу клинического исследования у пациентов, получавших препарат Иларис®, по сравнению с пациентами, получавшими триамцинолон, отмечалось увеличение средней активности АЛТ на 3,0 Ед/л и срединного значения активности АЛТ на 2,0 Ед/л, а также увеличение средней активности ACT на 2,7 Ед/л и срединного значения активности ACT на 2,0 Ед/л от исходного значения. Однако частота клинически значимого увеличения активности (>3 ВГН) ACT и АЛТ была более выражена у пациентов, получавших триамцинолон (на 2,5% для ACT и АЛТ), по сравнению с пациентами, получавшими терапию препаратом Иларис® (1,6% для АЛТ и 0,8% для ACT).
Триглицериды
На фоне терапии канакинумабом наблюдалось повышение концентрации триглицеридов в плазме крови в среднем на 33,5 мг/дл, а в группе применения триамцинолона – незначительное снижение на 3,1 мг/дл. Повышение концентрации триглицеридов более чем в 5 раз (по сравнению с ВГН) отмечалось в 2,4% случаев в группе применения канакинумаба и в 0,7% случаев в группе применения триамцинолона. Клиническая значимость данного наблюдения неизвестна.
Долгосрочные данные наблюдательного исследования
В рамках долгосрочного исследования по данным регистра выявлено 243 пациента с CAPS (85 педиатрических пациентов в возрасте от 2 до 17 лет и 158 взрослых пациентов старше 18 лет), которые получили лечение препаратом в рутинной клинической практике (в среднем в течение 3,8 лет). Профиль долгосрочной безопасности в данном исследовании соответствовал определенному в ходе интервенционных клинических исследований.
Педиатрические пациенты
В интервенционных клинических исследованиях препарат Иларис® получили 80 педиатрических пациентов с CAPS (в возрасте от 2 до 17 лет). В целом не отмечено клинически значимых различий безопасности и профиля переносимости препарата по сравнению с общей популяцией пациентов с CAPS (в целом 211 взрослых и педиатрических пациентов), включая общую частоту развития и тяжести эпизодов инфекций. Инфекции верхних дыхательных путей являлись наиболее часто отмечавшимися инфекционными заболеваниями.
В небольшом открытом клиническом исследовании дополнительно участвовали 6 пациентов в возрасте младше 2 лет. Профиль безопасности в данном исследовании соответствовал таковому для популяции в возрасте 2 лет и старше.
В 16-недельном исследовании препарат получили 102 пациента с TRAPS, H1DS/MKD и FMF в возрасте от 2 до 17 лет. В целом не отмечено клинически значимых различий безопасности и профиля переносимости препарата по сравнению с общей популяцией.
Применение у пациентов старше 65 лет
Не было выявлено различий профиля безопасности препарата у пациентов данной группы.
Если отмечено ухудшение клинического течения любого из указанных в инструкции побочных эффектов, или Вы заметили любые другие побочные эффекты, не указанные в инструкции, сообщите об этом врачу.
Передозировка
Полученные данные о передозировке препаратом ограничены. В клинических исследованиях на ранних этапах пациенты и здоровые добровольцы получали препарат внутривенно или п/к в дозе до 10 мг/кг, что не сопровождалось явлениями острой токсичности. При передозировке препаратом Иларис® следует обеспечить наблюдение пациента с целью выявления возможных нежелательных явлений, а также при необходимости немедленно начать соответствующую симптоматическую терапию.
Взаимодействие
Специальных исследований по взаимодействию препарата Иларис® с другими лекарственными препаратами не проводилось.
При применении одного из ингибиторов ИЛ-1 с ингибиторами фактора некроза опухоли (ФНО) отмечалось увеличение частоты развития серьезных инфекций. Применение препарата Иларис® с ингибиторами ФНО не рекомендовано в связи с увеличением риска развития серьезных инфекций.
Поскольку экспрессия изоферментов системы цитохрома Р450 в печени может быть подавлена цитокинами, стимулирующими хроническое воспаление, такими как ИЛ-1β, при применении мощных ингибиторов цитокинов, экспрессия изоферментов системы цитохрома Р450 в печени может быть нормализована. Это является клинически значимым для препаратов, метаболизирующихся с помощью изоферментов системы цитохрома Р450 и имеющих узкий терапевтический индекс, когда доза препарата подбирается индивидуально. При применении препарата Иларис® у пациентов, принимающих такие препараты, их дозу следует при необходимости корректировать (в зависимости от их клинического эффекта и концентрации действующего вещества в плазме крови).
В клинических исследованиях отмечено безопасное применение препарата Иларис® с противоподагрическими средствами.
Данные о влиянии живых вакцин, а также о возможном инфицировании у пациентов, получающим лечение препаратом, отсутствуют. Проводить вакцинацию живыми вакцинами у пациентов, получающих лечение препаратом Иларис®, следует только в случае, когда польза от вакцинации превышает возможный риск. Следует провести все рекомендованные местным календарем прививок вакцинации (включая пневмококковую вакцину и инактивированную вакцину против гриппа) до начала терапии препаратом Иларис®.
В случае необходимости вакцинацию живыми вакцинами проводят после начала терапии препаратом (по крайней мере через 3 месяца после последней инъекции препарата и за 3 месяца до следующей). Данные проведенных клинических исследований у взрослых здоровых добровольцев показывают, что однократное введение препарата Иларис® в дозе 300 мг не влияет на начало выработки антител и персистенцию поствакцинального ответа при вакцинации против гриппа и менингококковой инфекции (гликозилированной белковой вакциной).
В 56-недельном открытом клиническом исследовании у пациентов с CAPS в возрасте 4 лет и младше отмечено развитие достаточного количества антител у всех пациентов, получивших иммунизацию стандартными неживыми вакцинами.
Особые указания
Опыт применения препарата Иларис® у пациентов с CAPS без установленной мутации в NLRP3 гене ограничен.
Нейтропения и лейкопения
На фоне терапии ингибиторами ИЛ-1, включая препарат Иларис®, отмечено развитие нейтропении (абсолютное количество нейтрофилов ниже 1,5×109/л) и лейкопении. Не следует начинать лечение препаратом у пациентов с нейтропенией или лейкопенией. Перед началом терапии препаратом следует определить количество лейкоцитов, в том числе нейтрофилов и повторно оценить данные показатели через 1-2 месяца. В случае длительной или повторной терапии следует периодически контролировать данные показатели во время лечения. При выявлении снижения абсолютного количества нейтрофилов и лейкоцитов на фоне терапии препаратом следует обеспечить надлежащий контроль состояния пациента и при необходимости рассмотреть вопрос о прекращении лечения препаратом.
Злокачественные новообразования
Имеются сообщения о случаях развития злокачественных новообразований у пациентов, получающих препарат Иларис®, однако риск развития злокачественных новообразований на фоне терапии антителами, связывающими ИЛ-1, неизвестен.
Инфекционные заболевания
Применение препарата Иларис® может сопровождаться увеличением частоты развития серьезных инфекций, поэтому следует тщательно наблюдать пациента с целью выявления симптомов инфекции во время и после терапии препаратом. Следует соблюдать осторожность при применении препарата у пациентов с инфекциями, рецидивирующими инфекционными заболеваниями в анамнезе или сопутствующими заболеваниями, предрасполагающими к развитию инфекций. При CAPS, TRAPS, HIDS/MKB, FMF и болезни Стилла лечение препаратом Иларис® не следует начинать или продолжать у пациентов с активной инфекцией, требующей медицинского вмешательства. У пациентов с подагрическим артритом не следует начинать терапию препаратом в активный период инфекции. Не следует применять препарат одновременно с ингибиторами ФНО, так как это может приводить к увеличению риска развития серьезных инфекций.
При применении препарата Иларис® отмечались единичные случаи оппортунистических инфекций (в т.ч. аспергиллез, атипичные инфекционные заболевания микобактериальной этиологии, опоясывающий лишай). Невозможно исключить причинно-следственную связь между данными явлениями и применением препарата.
При применении препарата приблизительно у 12% пациентов с различными фенотипами CAPS наблюдались положительные результаты туберкулиновой пробы без каких-либо признаков туберкулезной инфекции (латентной или активной). Нет данных об увеличении риска реактивации туберкулезной инфекции при лечении моноклональными антителами к ИЛ-1 (например, препарат Иларис®). Перед применением препарата Иларис® необходимо обследовать всех пациентов с целью выявления активной или латентной туберкулезной инфекции (включая сбор анамнеза и проведение соответствующих скрининговых тестов, например, туберкулиновой пробы, тестов IGRA (Interferon-Gamma-Release-Assay) или рентгенологического обследования органов грудной клетки. Во время и после лечения препаратом следует тщательно контролировать состояние пациента с целью выявления туберкулезной инфекции. Следует информировать пациента о необходимости обращаться к врачу при появлении следующих симптомов на фоне и после терапии препаратом Иларис®: длительно сохраняющийся кашель, снижение массы тела, субфебрильная температура. В случае конверсии туберкулинового теста из негативного в позитивный, особенно у пациентов высокого риска, необходимо провести альтернативные скрининговые тесты. При выявлении туберкулезной инфекции лечение препаратом Иларис® не следует начинать или продолжать.
Синдром активации макрофагов у пациентов с болезнью Стилла у взрослых и сЮИА
Синдром активации макрофагов – известное жизнеугрожающее состояние, которое может развиваться у пациентов с ревматическими заболеваниями, в частности у пациентов с болезнью Стилла, и требовать интенсивной терапии. Врачу следует внимательно относиться к симптомам инфекции или ухудшению течения заболевания, известными как пусковой механизм для синдрома активации макрофагов. Данные клинических исследований указывают, что препарат Иларис®, по всей вероятности, не увеличивает риск развития синдрома активации макрофагов у пациентов с сЮИА, однако, не позволяют сделать окончательных выводов.
Реакции гиперчувствительности
При применении препарата Иларис® сообщалось о реакциях гиперчувствительности. В большинстве случаев данные реакции были выражены в легкой степени. При применении препарата не наблюдалось развития анафилактоидных или анафилактических реакций. Однако при применении препарата Иларис® нельзя исключить риск развития тяжелых реакций гиперчувствительности, которые могут отмечаться при инъекционном введении препаратов белкового происхождения.
Нарушение функции печени
В клинических исследованиях отмечены транзиторное повышение активности печеночных трансаминаз или концентрации билирубина.
Влияние на способность управлять транспортными средствами, механизмами
Пациентам, у которых на фоне применения препарата Иларис® возникает вертиго, следует воздержаться от управления транспортными средствами или механизмами до полного исчезновения данного нежелательного явления.
Форма выпуска
По 1 мл раствора для подкожного введения 150 мг/мл во флакон из бесцветного стекла гидролитического класса I вместимостью 2 мл, укупоренный пробкой из хлорбутилового каучука, ламинированного фторполимером, обкатанной алюминиевым колпачком с пластиковой отщелкивающейся крышкой.
По 1 флакону вместе с инструкцией по медицинскому применению в пачку картонную. Условия ХРАНЕНИЯ
Хранить в защищенном от света месте при температуре от 2 до 8 °С. Не замораживать.
Хранить в оригинальной картонной пачке.
Препарат следует хранить в недоступном для детей месте.
Срок годности
3 года.
Препарат не применять по истечении срока годности.
Условия отпуска
По рецепту.
Производитель
Новартис Фарма Штейн АГ, Шаффхаузерштрассе, 4332 Штейн, Швейцария/ Novartis Pharma Stein AG, Schaffhauserstrasse, 4332 Stein, Switzerland.
Характеристики

Цены в аптеках на Иларис

История стоимости Иларис
Цены Иларис и наличие в аптеках в Тамбове
| Список аптек | Адрес | Часы работы | Цена |
|---|---|---|---|
| 08:00-20:00 Пн-Вс | 652 151 ₽ | ||
| 08:00-20:00 Пн-Вс | 652 151 ₽ | ||
| 08:00-22:00 Пн-Вс | 652 151 ₽ | ||
| 08:00-22:00 Пн-Вс | 652 151 ₽ | ||
| 08:00-20:00 Пн-Вс | 652 151 ₽ |

Отзывы о Иларис





Часто задаваемые вопросы
По какой цене можно купить Иларис?
Какая страна производства препарата Иларис?
Какой срок годности Иларис?
Как хранить Иларис?








Дистанционная торговля лекарственными препаратами осуществляется исключительно аптечными организациями, имеющими действующую лицензию на фармацевтическую деятельность, а также разрешение на дистанционную торговлю лекарственными препаратами. Дистанционная торговля рецептурными лекарственными препаратами, наркотическими и психотропными, а также спиртосодержащими лекарственными препаратами запрещена действующим законодательством РФ и не осуществляется.
На информационном ресурсе применяются рекомендательные технологии .